Introducción
Los tumores intracraneales en pacientes de 0-19 años presentan una incidencia de 5,81 casos por cada 100.000 individuos1, siendo la segunda patología maligna más frecuente en este grupo de edad2. A pesar de ser tres veces menos comunes que los tumores intracraneales en adultos, son los tumores sólidos más frecuentes en pediatría y la principal causa de muerte por cáncer en esta población3. En los primeros años de vida hay mayor incidencia de tumores de origen embrionario, como el meduloblastoma y el tumor teratoide/rabdoide atípico, mientras que en niños de mayor edad prevalecen los tumores de origen glial2. La localización en la infancia suele ser infratentorial, excepto en el caso de los tumores congénitos y en menores de 1 año, que son más frecuentes en localización supratentorial, que se asocia a histopatología benigna4,5.
La morbimortalidad de la patología tumoral cerebral está dada no solo por la presencia de un tumor expansivo intracraneal, sino también por su tratamiento, sea este la resección quirúrgica o quimioterapia/radioterapia, llevando a graves daños cognitivos y del desarrollo neurológico a largo plazo; incluso, la radioterapia puede derivar en la aparición de tumores secundarios3. Esto presenta un gran desafío al momento de tomar una conducta terapéutica.
El primer estudio por imágenes que se realiza en pacientes pediátricos con síntomas neurológicos que sugieren origen tumoral suele ser la tomografía computada6. Para la caracterización de estas lesiones, así como para la planificación del tratamiento a seguir, se utiliza la resonancia magnética (RM) con contraste intravenoso.
El propósito de este estudio es hacer un análisis de la incidencia y el comportamiento de los tumores intracraneales en individuos menores de 1 año, teniendo en cuenta la sobrevida posterior al tratamiento seleccionado y la comorbilidad consecuente.
Método
Se realizó un estudio de cohorte retrospectivo teniendo en cuenta pacientes de hasta 12 meses de vida con diagnóstico por RM de tumor primario intracraneal, que hubieran realizado estudios por imágenes en la institución. Se excluyeron los pacientes con tumores orbitarios y faciales, lesiones quísticas y de origen vascular, y los que no contaban con RM previa a la cirugía.
Para el análisis se obtuvo información de la historia clínica electrónica de los pacientes; los datos que se tuvieron en cuenta fueron la edad y el sexo de los pacientes, la topografía, la localización, la histopatología y el comportamiento del tumor, la evolución de los pacientes tanto tratados como no tratados, el tratamiento recibido en aquellos que se trataron, la progresión de la enfermedad, la sobrevida a 5 años, los años libres de enfermedad, y la tasa de muerte pre- y postratamiento.
Entre las características radiológicas se registraron la conservación de la línea media, la hemorragia asociada, el patrón de realce poscontraste, la hidrocefalia y la diseminación al raquis o las leptomeninges.
Las variables continuas se describieron mediante media y desvío estándar. Las variables continuas fueron evaluadas con la prueba U de Mann Whitney, y las variables categóricas con la de chi al cuadrado. El análisis estadístico se realizó mediante el software SPSS versión 21.0. El límite para la relevancia estadística se estableció en p = 0,05.
El estudio fue aprobado por el comité de ética institucional. Toda la información personal de los pacientes fue manejada en forma confidencial, conforme a la Ley Nacional de Protección de Datos Personales 25.326 (Ley de Habeas Data).
Resultados
Entre los años 2010 y 2022, un total de 11 pacientes menores de 1 año se realizaron RM con hallazgos sugestivos de tumor primario intracraneal. Cuatro fueron de sexo masculino y siete de sexo femenino. El 63,6% fueron diagnosticados antes de cumplir los 6 meses de vida, mientras que el 36,4% iniciaron con síntomas entre los 6 y 12 meses. La media de edad al momento del diagnóstico fue de 4,45 meses.
El tipo histológico más frecuente fue el glial, hallado en seis pacientes. Dentro de los tumores de alto grado se encontraron dos tumores teratoides rabdoides atípicos, asociados a peor pronóstico ya que ambos pacientes fallecieron, y un ependimoma anaplásico. De los 11 pacientes, siete tuvieron anatomía patológica que confirmaba el diagnóstico y cuatro se diagnosticaron exclusivamente por sus características radiológicas
Encontramos seis pacientes con tumores a nivel supratentorial y cinco a nivel infratentorial. De los tumores supratentoriales, tres se encontraban en la línea media, dos a nivel lobar y uno intraventricular.
Del total de los tumores, siete restringían en secuencia de difusión, nueve reforzaban tras la administración de contraste intravenoso y dos presentaban diseminación a las leptomeninges. Se observó desviación de la línea media en ocho pacientes e hidrocefalia en seis de ellos.
Las características radiológicas de todos los pacientes se describen en la tabla 1.
Tabla 1. Cohorte de pacientes incluidos en este trabajo
| Paciente | Histología | Edad (meses) | Sexo | Topografía | Ubicación | Línea media | Restricción | Realce | Patrón refuerzo | Hemorragia | Hidrocefalia | Diseminación al raquis | Diseminación leptomeníngea |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Tumor teratoide rabdoide atípico | 1 | F | Infratentorial | Línea media | Sí | Sí | Sí | Masa sólida, heterogéneo | Sí | Sí | No | No |
| 2 | Tumor teratoide rabdoide atípico | > 6 | M | Infratentorial | Tronco del encéfalo | Sí | Sí | Sí | Masa sólido-quística, heterogéneo | No | No | No | Sí |
| 3 | Ependimoma anaplásico | 11 | F | Infratentorial | Tronco del encéfalo | Sí | Sí | Sí | Heterogéneo | Hemorragia vs. vasos | Sí | No | No |
| 4 | Glioma con anaplasia focal y cinética elevada por KI67 | 3 | M | Supratentorial | Línea media | Sí | No | Sí | Masa sólida, homogéneo | No | Sí | Sí | Sí |
| 5 | Astrocitoma pilomixoide | 5 | F | Supratentorial | Línea media | Sí | No | Sí | Masa sólida, homogéneo | No | No | No | No |
| 6 | Astrocitoma óptico-quiasmático | 2 | M | Supratentorial | Línea media | Sí | Sí | Sí | Parcial | No | Sí | No | No |
| 7 | Glioma de bajo grado | > 6 | M | Infratentorial | Tronco del encéfalo | No | Sí | No | – | Sí | Sí | No | No |
| 8 | Tumor de células claras | 1 | M | Infratentorial | Tronco del encéfalo | No | No | Sí | Heterogéneo | Sí | No | No | No |
| 9 | Astrocitoma desmoplásico infantil | 4 | F | Supratentorial | Lobar | Sí | No | Sí | Heterogéneo | No | Sí | No | No |
| 10 | Astrocitoma gigantocelular | 0 | F | Supratentorial | Intraventricular | SI | Sí | No | – | No | No | No | No |
| 11 | Tumor neuronal de bajo grado | 10 | F | Supratentorial | Lobar | No | Sí | Sí | Nodular | Hemorragia vs. calcificación | No | No | No |
El síntoma más frecuente fueron los vómitos (tres pacientes) y el signo más frecuente fue el aumento del perímetro cefálico (tres pacientes). Dos pacientes presentaron parálisis facial y el resto manifestaron síntomas como alteración de la apertura ocular, nistagmo y epilepsia; uno de ellos no presentó síntomas neurológicos, sino que fue un hallazgo en el contexto de diagnóstico de esclerosis tuberosa.
De los 11 pacientes estudiados, solo cinco recibieron tratamiento quirúrgico, de los cuales dos fallecieron durante el procedimiento y uno falleció posterior al mismo; de los dos pacientes que sobrevivieron, uno presentó progresión de la enfermedad y el otro no. No encontramos diferencias en cuanto a la ubicación supra- o infratentorial entre los pacientes que fallecieron y los que no (p = 0,24). Observamos una mayor frecuencia de lesiones con refuerzo poscontraste en los pacientes que fallecieron en el seguimiento (p = 0,03). En cuanto a los pacientes no intervenidos quirúrgicamente, tres recibieron tratamiento quimioterápico, con progresión de la enfermedad en dos de ellos, uno no recibió ningún tipo de tratamiento y se mantuvo con conducta expectante debido a que no presentó signos ni síntomas neurológicos, y otro recibió tratamiento para resolver la hidrocefalia con colocación de una válvula de derivación ventriculoperitoneal sin progresión de la enfermedad. El paciente restante no continuó seguimiento en nuestra institución, por lo que no contamos con información actualizada de su estado de salud.
En los siete pacientes que actualmente siguen con vida y pudo hacerse seguimiento, se evaluó la progresión de la enfermedad según el comportamiento por imágenes, así como la comorbilidad tanto postratamiento como la propia de la evolución del tumor. Tres pacientes presentan desarrollo neurológico normal, de los cuales solo uno recibió tratamiento de la lesión con resección parcial, mientras que en los dos restantes se colocó derivación ventriculoperitoneal a uno y se tomó una conducta expectante en el otro. De los cuatro pacientes que sí presentaron comorbilidad, dos persisten con trastornos en la deglución habiendo recibido como único tratamiento quimioterapia, uno presenta exotropía del ojo izquierdo y otro manifiesta daño neurológico con retraso madurativo, insuficiencia hipofisaria y alteraciones visuales.
Discusión
Presentamos una serie de casos de tumores intracraneales en pacientes menores de 1 año con diagnóstico imagenológico y patológico, y seguimiento a largo plazo.
Aproximadamente el 63% de todos los tumores en pacientes menores de 19 años derivan de células gliales, como los astrocitomas (36%); sin embargo, en los primeros 12 meses de vida, al menos el 75% de los tumores intracraneales son de origen neuroepitelial5, mientras que los tumores intracraneales congénitos son principalmente derivados de células tumorales germinales, siendo el teratoma el tipo histológico más común en pacientes menores de 2 meses de vida4.
En nuestro estudio se vio que, a pesar de que la población estudiada se encontraba dentro de los primeros 12 meses de vida, el tipo histológico más frecuente fue el glial, correspondiendo al 54,5%, de los cuales dos fueron gliomas y cuatro fueron astrocíticos. Ningún paciente presentó tumores derivados de células germinales y solo dos fueron histológicamente embrionarios (tumores teratoides rabdoides atípicos ambos). Un tercer tumor de alto grado presentó histopatología correspondiente a ependimoma anaplásico. Un paciente presentó anatomía patológica acorde a un tumor de células claras, y por último uno de ellos no tuvo anatomía patológica pero las características imagenológicas sugerían un tumor de bajo grado.
Durante el periodo neonatal, así como durante los primeros 2 años de vida, predomina la localización supratentorial6, lo cual coincide con lo detectado en nuestra muestra.
La presentación clínica varía dependiendo de la localización del tumor y la edad del paciente. Durante los primeros años de vida, la presentación suele ser inespecífica, con signos advertidos por los padres, lo cual lleva a una demora en el diagnóstico. En edades más avanzadas suelen presentarse signos y síntomas más claros, como convulsiones, déficit neurológico localizado, cefalea, vómitos y compromiso de pares craneales2. En la población estudiada, los principales motivos de consulta fueron aumento del perímetro cefálico y vómitos (45,5%); dos pacientes iniciaron con parálisis facial, uno con nistagmo, uno con dificultad para la apertura ocular y solo uno presentó convulsiones. Hubo un caso de hallazgo incidental en el contexto del diagnóstico de esclerosis tuberosa, persistiendo el paciente asintomático hasta la fecha.
Dentro de los tumores embrionarios, los más frecuentes son el meduloblastoma y el tumor teratoide rabdoide atípico. Ambos tienen un comportamiento similar en la RM dada su alta celularidad, viéndose en la fosa posterior una masa heterogénea debido a la presencia de hemorragia, quistes y calcificaciones. Son iso- o hipointensos en la secuencia ponderada en T1 (con focos de alta señal en caso de hemorragia) e hipointensos en T2, y presentan marcada restricción en difusión. El meduloblastoma tiene una incidencia del 30-40% dentro de los tumores de fosa posterior, con un pico a los 3-4 años y otro a los 7-10 años, lo cual coincide con la ausencia de este tumor en la población estudiada. El tumor teratoide rabdoide atípico de fosa posterior tiene una incidencia menor del 5% en la población pediátrica, pero dentro de los primeros 2 años de vida alcanza un 25%. En un 50% de los casos se ubican en el ángulo pontocerebeloso. Al momento del diagnóstico, un 10-30% de los pacientes presentan diseminación7. El tratamiento suele ser multimodal, con una supervivencia a 5 años del 30% y peor pronóstico en los pacientes menores de 6 meses en el momento del diagnóstico3. En nuestro estudio, dos pacientes de 1 y 6 meses de vida presentaron tumor teratoide rabdoide atípico, encontrándose ambos en la fosa posterior, pero ninguno de ellos en el ángulo pontocerebeloso (APC), sino que se localizaron en el hemisferio cerebeloso izquierdo uno y en la línea media el otro. Presentaban las características imagenológicas descritas en la literatura (Fig. 1) y uno de ellos se asoció a diseminación leptomeníngea. Ambos pacientes recibieron tratamiento quirúrgico; uno murió durante el procedimiento y el otro en el periodo posquirúrgico.
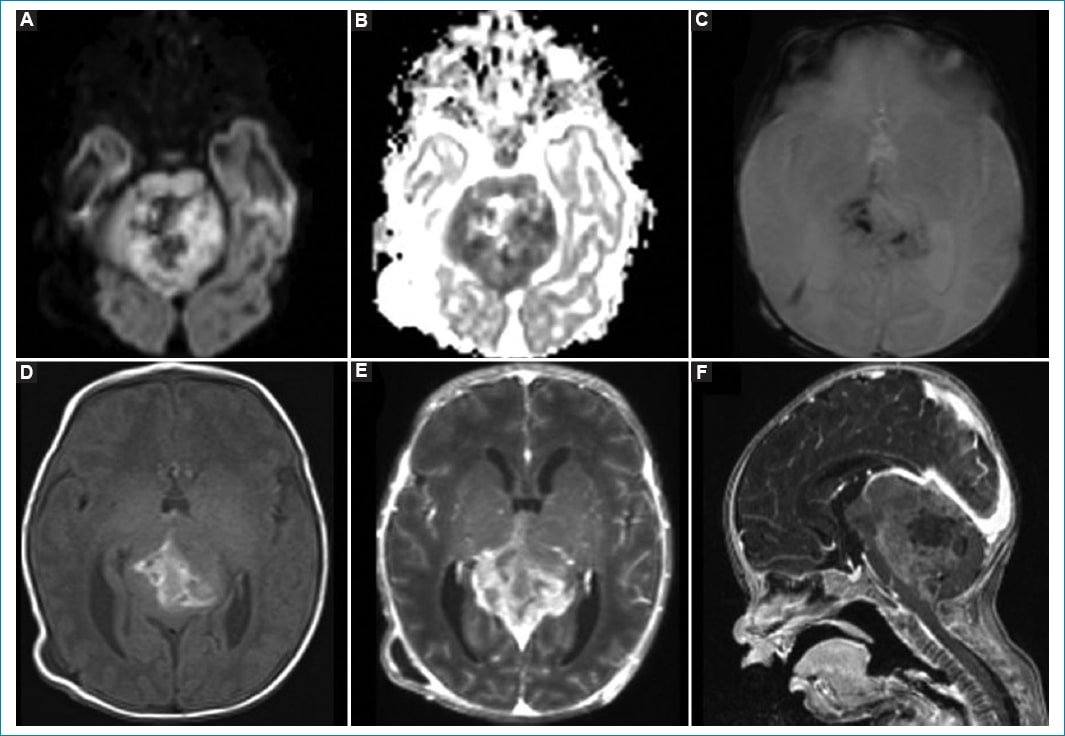
Figura 1. Tumor teratoide rabdoide atípico en un paciente de 1 mes de vida. Lesión expansiva, heterogénea, con restricción de las áreas sólidas en la secuencia de difusión (A) con representación en el mapa de ADC (B). En la secuencia de susceptibilidad magnética (C) se observan áreas de baja señal que coinciden con áreas de alta señal en T1 (D), sugestivo de hemorragia intralesional. Tras la administración de contraste intravenoso (E y F) se observa realce vívido y heterogéneo.
Los tumores de origen ependimario son el tercer tipo de tumor primario cerebral en pediatría, con un pico dentro de los primeros 4 años de vida y predominancia en el sexo masculino. Son más frecuentes en localización infratentorial, representando el 10-15% de los tumores de la fosa posterior7. Actualmente se clasifican, según su localización, en cinco grupos: ependimomas supratentoriales, ependimomas de fosa posterior grupos A y B, ependimomas medulares, ependimomas mixopapilares y subependimomas8. En nuestro estudio encontramos un solo caso de este tipo histológico, ubicado en la fosa posterior y con características propias de un tumor de grado 3. Se consideran de grado 3 aquellos con alta celularidad, atipia nuclear, hipercromatismo, marcada actividad mitótica y necrosis o proliferación vascular asociada. En los estudios por imágenes se encontrarán lesiones en la fosa posterior que nacen del piso del IV ventrículo, con extensión a los forámenes de Luschka y Magendie. Los ependimomas de fosa posterior del grupo A nacen principalmente del receso lateral del IV ventrículo, y tienen menores posibilidades de ser resecados que los del grupo B, que suelen nacer en la línea media. En la RM habitualmente presentan señal baja ponderada en T1 y heterogénea en T2, dado que en torno al 50% presentan calcificaciones y el 10% presentan hemorragia. En difusión muestran restricción intermedia, y el realce tras la administración de contraste intravenoso es variable en intensidad1. En nuestro paciente, el realce fue marcadamente heterogéneo debido a las múltiples áreas sólido-quísticas y focos de hemorragia (Fig. 2). Con independencia del grado de resecabilidad, en tumores considerados de grado 3 el tratamiento de elección es la radiación, viéndose mejores resultados cuando a esta le sigue la quimioterapia; en algunos pacientes se prefiere evitar la radiación y realizar solo quimioterapia debido a los efectos adversos en edades tempranas7. En nuestro paciente se decidió tratamiento únicamente con quimioterapia, sin evidencia de progresión de la enfermedad al día de hoy, pero con secuelas neurológicas como trastorno de la deglución y de la marcha.
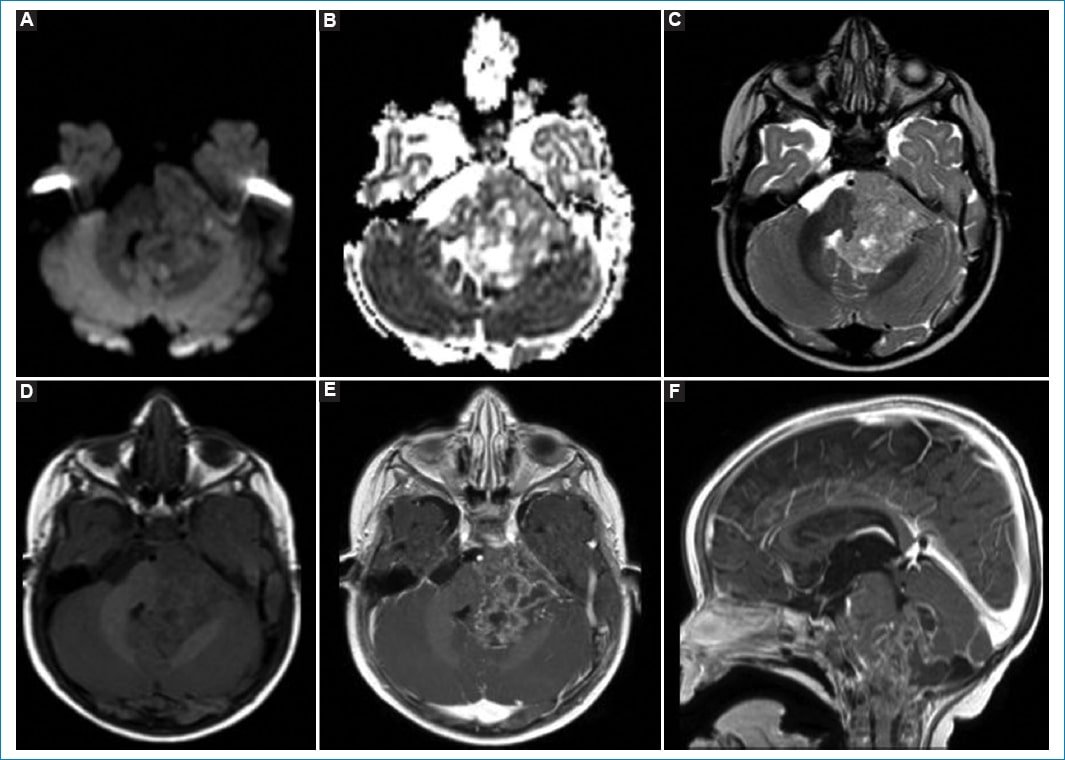
Figura 2. Ependimoma de fosa posterior de grado 3 en un paciente de 11 meses de vida. En las secuencias de difusión (A) y ADC (B) se observa una lesión expansiva en el IV ventrículo, que se extiende hacia posterior, lateral y anterior, sin restricción. Presenta señal heterogénea en la secuencia ponderada en T2 (C) y T1 (D) con restos hemáticos y áreas quísticas. Tras la administración de contraste (E) presenta realce heterogéneo, principalmente en la porción sólida. En cortes sagitales (F) se observa la extensión de la misma hacia superior e inferior a través de los forámenes de Magendie y Luschka.
En cuanto a los tumores de origen glial, los más frecuentes en la población analizada fueron los astrocitomas, viéndose cuatro casos de bajo grado que comprendieron un astrocitoma pilomixoide, un astrocitoma óptico-quiasmático, un astrocitoma desmoplásico infantil y un astrocitoma gigantocelular. Los gliomas de bajo grado (grados 1 y 2) son los tumores del sistema nervioso central más frecuentes en pacientes pediátricos, representando el astrocitoma pilocítico el 15% de todos los tumores intracraneales pediátricos. En nuestro estudio se halló una variante de astrocitoma pilocítico, el astrocitoma pilomixoide, con una edad promedio de diagnóstico a los 18 meses y un comportamiento benigno en la RM, ya que se presentan como lesiones bien circunscritas, sin edema periférico ni infiltración parenquimatosa, y con un leve componente quístico y realce homogéneo tras la administración de contraste9; características que coinciden con nuestros hallazgos (Fig. 3). Si bien el tratamiento de elección es la resección quirúrgica, con pronóstico más favorable cuanto mayor es el volumen tumoral resecado, en nuestro paciente se realizó el procedimiento quirúrgico y falleció durante el mismo. En los otros tres casos de astrocitomas que observamos, las características radiológicas fueron similares: lesiones sin hemorragia y sin diseminación al momento del diagnóstico, con realce intermedio o nulo tras la administración de contraste intravenoso y con restricción variable en la secuencia de difusión. El paciente con astrocitoma óptico quiasmático recibió quimioterapia, con posterior progresión de la enfermedad y trastornos de la deglución como secuela en la actualidad. En el paciente con astrocitoma desmoplásico infantil se realizó resección parcial y en el paciente con astrocitoma gigantocelular se tomó una conducta expectante; ambos están sin secuelas actualmente.
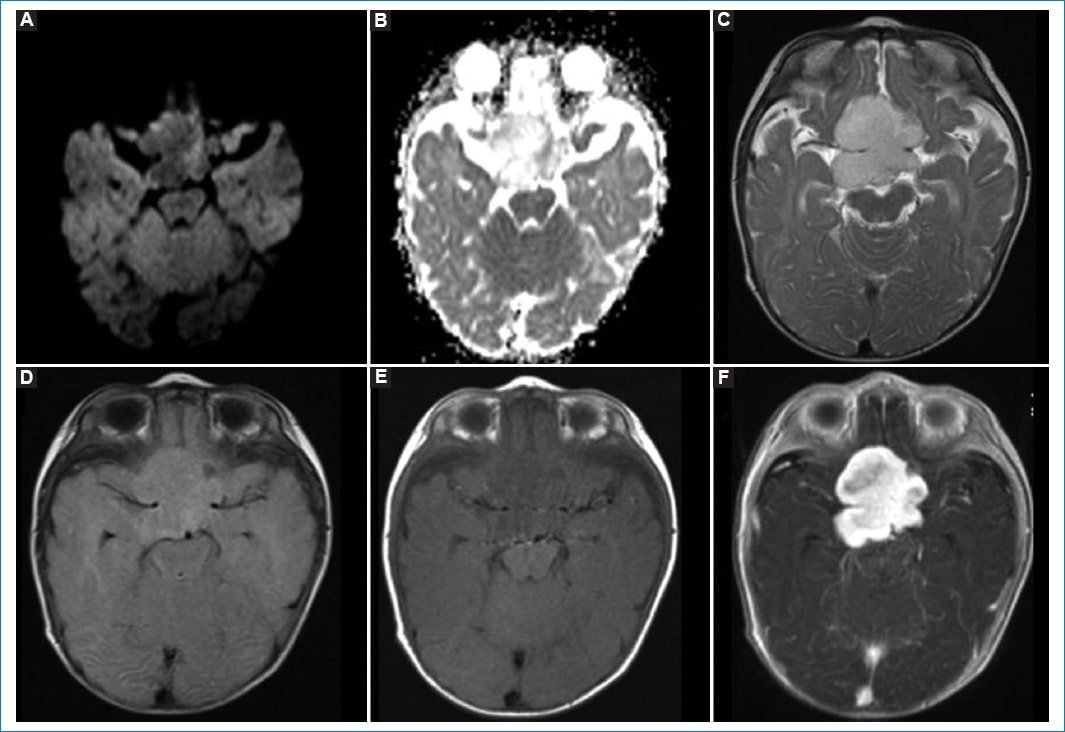
Figura 3. Astrocitoma pilomixoide en un paciente de 5 meses de vida. Se observa una imagen selar-supraselar de aspecto polilobulado, sin restricción en la secuencia de difusión (A) ni caída en el mapa de ADC (B). Presenta señal alta homogénea en T2 (C) y FLAIR (D), y baja ponderada en T1 (E). Tras la administración de contraste intravenoso (F) presenta realce intenso, principalmente homogéneo, a excepción de algunos sectores anteriores y periféricos. Rodea y desplaza las estructuras vasculares.
Este estudio presenta dos grandes limitaciones. Por un lado, al realizar un análisis retrospectivo no se pudo hacer un seguimiento detallado de los pacientes elegidos y solo contamos con la información plasmada en la historia clínica electrónica. En un caso perdimos por completo el seguimiento dado que el paciente dejó de atenderse en nuestro centro. Por otro lado, el número de pacientes es mucho menor que el esperado, por lo que los resultados podrían no ser extrapolables a la población general.
Conclusión
Si bien contamos con un número reducido de pacientes para analizar el comportamiento de los tumores intracraneales en pacientes dentro del primer año de vida, este estudio demuestra que algunas características imagenológicas pueden ayudar a estimar el pronóstico de las lesiones neoplásicas en este grupo etario.
Financiamiento
Los autores declaran no haber recibido financiamiento para este estudio.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Responsabilidades éticas
Protección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores han obtenido la aprobación del Comité de Ética para el análisis y publicación de datos clínicos obtenidos de forma rutinaria. El consentimiento informado de los pacientes no fue requerido por tratarse de un estudio observacional retrospectivo.
Uso de inteligencia artificial para generar textos. Los autores declaran que no han utilizado ningún tipo de inteligencia artificial generativa en la redacción de este manuscrito ni para la creación de figuras, gráficos, tablas o sus correspondientes pies o leyendas.
